
phylo-node
v0.1.4
Published
phylo-node: A Molecular Phylogenetic Toolkit using Node.js
Downloads
74
Maintainers
 dohalloran
dohalloranReadme
phylo-node


![]()
![]()

![]()
phylo-node: A Molecular Phylogenetic Toolkit using Node.js

CONTENTS
Click on Demo to get full length video
Getting Started
Install the module with:
npm install phylo-nodeUsage
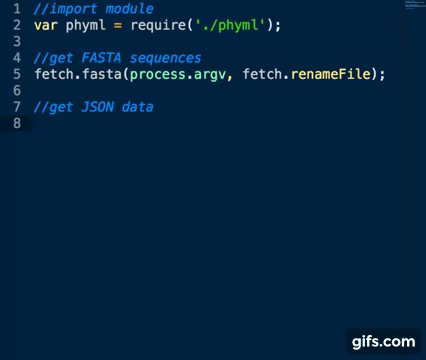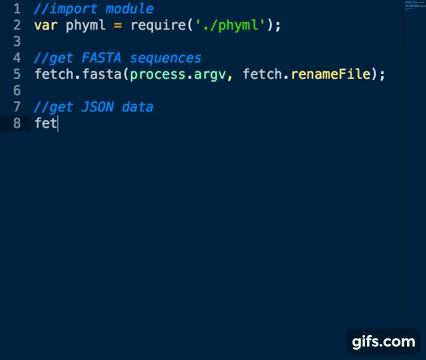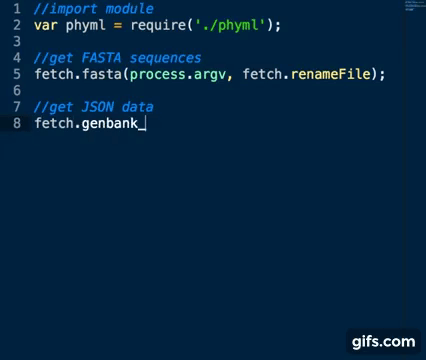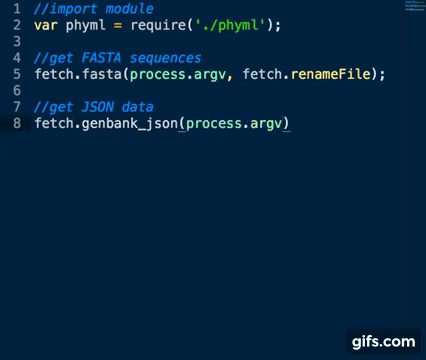
Require module, for example:
var phyml = require('./phyml')ensure executables are in your $PATH
Get FASTA Sequences
Sequence Accession Numbers are collected from the commandline separated by a space (not a comma)
Node uses NCBI e-utilities to download sequences in fastA format:
var fetch = require('./fetch_seqs')
fetch.fasta(process.argv, fetch.renameFile)Basic usage: node app.js inputfile [list of space separated accession numbers]
node app.js NM_001028053.2 AF032112.1Get Sequence information in ASN.1 format
Sequence Accession Numbers are collected as per fastA sequences above using the genbank_json method:
var fetch = require('./fetch_seqs')
fetch.genbank_json(process.argv)Basic usage: node app.js inputfile [list of space separated accession numbers]
node app.js NM_001028053.2 AF032112.1Download executables
Download executable files:
var get_executable = require('./get_executable.js')
get_executable.software(process.argv[2])Basic usage: node app.js URL
node app.js http://www.clustal.org/omega/clustalo-1.2.2-Ubuntu-x86_64Note: objects for other tools i.e. PhyML, Clustal Omega, and MUSCLE contain their own methods for downloading binaries (see below)
Server
Create a web server:
Basic usage:
node http_server.js
Node server listening on port 8080Point browser to localhost:8080
Note: to create a JBrowse server, it should be downloaded and configured as per the developer guidelines described here
Bowtie2
Run Bowtie2 program
var base = require('../../../Wrapper_Core/base-wrap')
var bowtie2 = require('./bowtie2')
base.call_(process.argv[2], process.argv[3], process.argv, bowtie2.run_)Basic usage: node app.js index-file -U fastQ-reads
node app.js ../../../Input_examples/index_elegans/c_elegans -U ../../../Input_examples/reads.fqTrimmomatic
Run Trimmomatic program
var base = require('../../../Wrapper_Core/base-wrap')
var trimmomatic = require('./trimmomatic')
base.call_(process.argv[2], process.argv[3], process.argv, trimmomatic.run_)Basic usage: node app.js path-to-jar input-file [insert any flags (from flags below)]
node app.js /usr/share/java/trimmomatic.jar ../../../Input_examples/reads.fq Output/outsy.fq -phred33 ILLUMINACLIP:TruSeq3-SE:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36| FLAG | DETAILS |
| --------------------- |:------------------------------------------------------------------:|
| ILLUMINACLIP | Cut adapter and other illumina-specific sequences from the read |
| SLIDINGWINDOW | Perform a sliding window trimming |
| LEADING | Cut bases off the start of a read, if below a threshold quality |
| TRAILING | Cut bases off the end of a read, if below a threshold quality |
| CROP | Cut the read to a specified length |
| HEADCROP | Cut the specified number of bases from the start of the read |
| MINLEN | Drop the read if it is below a specified length |
| TOPHRED33 | Convert quality scores to Phred-33 |
| TOPHRED64 | Convert quality scores to Phred-64 |
Note: must have Java Runtime environment and Trimmomatic jar
PhyML
Download PhyML using this command
var phyml = require('./phyml.js')
phyml.getphyml()Run phyml program
var base = require('../../../Wrapper_Core/base-wrap')
var phyml = require('./phyml.js')
var outFile = './Output/PhyML.txt'
base.call_(process.argv[2], outFile, process.argv, phyml.run_)Basic usage: node app.js inputfile [insert any flags (from flags below)]
node app.js example_PhyML.phy -q -d aa -m JTT -c 4 -a e| FLAG | FIELD |
| --------------------- |:---------------------------------------:|
| -d | data_type |
| -q | |
| -n | nb_data_sets |
| -b | int |
| -m | model |
| -f | e | d | 'fA fC fG fT' |
| -t | ts/tv_ratio |
| -v | prop_invar |
| -c | nb_subst_cat |
| -a | gamma |
| -s | move |
| -u | user_tree_file |
| -o | 'tlr' | 'tl' | 'tr' | 'l' | 'r' | 'n' |
| --rand_start | |
| --n_rand_starts | num |
| --r_seed | num |
| --print_site_lnl | |
| --print_trace | |
Primer3
Run Primer3 program
var base = require('../../../Wrapper_Core/base-wrap')
var primer3 = require('./primer3.js')
var outFile = './Output/primer3.txt'
base.call_(process.argv[2], outFile, process.argv, primer3.run_)Basic usage: node app.js filename [-flags (from table below)]
node app.js example_p3 -format_output| FLAGS |
| ------------------------------------- |
| -format_output |
| -default_version=1|-default_version=2 |
| -io_version=4 |
| -p3_settings_file= file_path |
| -echo_settings_file |
| -strict_tags |
| -output= file_path |
| -error= file_path |
MUSCLE
Download muscle executable
var muscle = require('./muscle.js')
muscle.getmuscle()Run MUSCLE program
var base = require('../../../Wrapper_Core/base-wrap')
var muscle = require('./muscle.js')
var outFile = './Output/Muscle_Result.aln'
base.call_(process.argv[2], outFile, process.argv, muscle.run_)Basic usage: node app.js inputfile [insert any flags preceeded by '-' sign and seperated by a space (from flags below)]
node app.js DNA.fasta -msf -html| FLAG | FUNCTION | | ------------- |:----------------------------------------------:| | -diags | Find diagonals (faster for similar sequences) | | -html | Write output in HTML format (default FASTA) | | -msf | Write output in GCG MSF format (default FASTA) | | -clw | Write output in CLUSTALW format (default FASTA)| | -clwstrict | As -clw, with 'CLUSTAL W (1.81)' header | | -quiet | Do not write progress messages to stderr |
Clustal Omega
Download Clustal Omega executable
var clustal_Omega = require('./clustal_Omega.js')
clustal_Omega.getclustal()Run Clustal Omega program
var base = require('../../../Wrapper_Core/base-wrap')
var clustal_Omega = require('./clustal_Omega.js')
var outFile = './Output/Clustal_Result.aln'
base.call_(process.argv[2], outFile, process.argv, clustal_Omega.run_)Basic usage: node app.js inputfile [insert any flags preceeded by '--' sign and seperated by a space]
node app.js DNA.fasta --outfmt phy| FLAG | FUNCTION | | -------------------------- |:--------------------------------------------------------------------------------------:| | --full | Use full distance matrix for guide-tree calculation (slow; mBed is default) | | --full-iter | Use full distance matrix for guide-tree calculation during iteration (mBed is default) | | --cluster-size | Write output in GCG MSF format (default FASTA) | | --use-kimura | use Kimura distance correction for aligned sequences (default no) | | --percent-id | convert distances into percent identities (default no) | | --outfmt | {a2m=fa[sta],clu[stal],msf,phy[lip],selex,st[ockholm],vie[nna]} | | --resno | in Clustal format print residue numbers (default no) | | --wrap | number of residues before line-wrap in output | | --output-order | {input-order,tree-order} | | --iter | Number of (combined guide tree/HMM) iterations | | --max-guidetree-iterations | Maximum guide tree iterations | | --max-hmm-iterations | Maximum number of HMM iterations |
Kalign
Run Kalign program
var base = require('../../../Wrapper_Core/base-wrap')
var kalign = require('./kalign.js')
var outFile = './Output/kalign_Result.aln'
base.call_(process.argv[2], outFile, process.argv, kalign.run_)Basic usage: node app.js inputfile [insert any flags preceeded by '-' sign and seperated by a space]
node app.js DNA.fasta -gpo -f | FLAG | FUNCTION | | -------- |:-----------------------------------------------------------------------------:| | -gpo | Gap open penalty (default 6.0). | | -gpe | Gap extension penalty (default 0.9). | | -p | Wu-Manber algorithm used in both distance calculation and dynamic programming | | -w | Wu-Manber algorithm not used at all | | -f | fast heuristic alignment | | -q | 'quiet' - no messages are sent to standard error |
PAL2NAL
Run PAL2NAL program
var base = require('../../../Wrapper_Core/base-wrap')
var pal2nal = require('./pal2nal.js')
var outFile = './Output/result.codon'
base.call_(process.argv[2], outFile, process.argv, pal2nal.run_)Basic usage: node app.js input.aln input.fasta [insert any flags from below]
node app.js DNA.aln DNA.fasta -nomismatch | FLAG | FUNCTION | | ----------- |:-------------------------------------------------------------------------------:| | -h | show help | | -blockonly | Show only user specified blocks | | -output | (clustal,paml,fasta,codon) Output format, default = clustal | | -nogap | remove columns with gaps and inframe stop codons | | -nomismatch | remove mismatched codons (mismatch between pep and cDNA) from the output | | -codontable | 1 (default),2,3,4,5,6,9,10,11,12,13,14,15,16,21,22,23 NCBI GenBank codon table | | -html | HTML output (only for the web server) | | -nostderr | No STDERR messages (only for the web server) |
Note: must have Perl installed
Slr
Run Slr program
var base = require('../../../Wrapper_Core/base-wrap')
var Slr = require('./Slr.js')
var outFile = './Output/Slr_Results.txt'
base.call_(process.argv[2], outFile, process.argv, Slr.run_)Basic usage: node app.js input.paml input.trees [insert any flags from below]
node app.js bglobin.paml bglobin.trees timemem 1 | FLAG | FUNCTION | | ----------------|:------------------------------------------------------:| | -reoptimise | 0 (no), 1(yes), 2(set branch lengths to random values) | | -kappa | value for kappa | | -omega | Value for omega (dN/dS) | | -branopt | 0: fixed, 1: optimise, 2: proportional | | -codonf | 0: F61/F60 1: F3x4 2: F1x4 | | -freqtype | 0, 1, 2, 3 | | -positive_only | 0(no) or 1(yes) | | -nucleof | 0: none, 1: adjust by a constant N_{ab}. | | -aminof | 0(constant), 1, 2 | | -freqtype | 0, 1, 2, 3 | | -timemem | summary of real time and CPU time used 1:yes 0:no | | -skipsitewise | Skip sitewise estimation of omega |
Codeml
Run Codeml program
var base = require('../../../Wrapper_Core/base-wrap')
var codeml = require('./codeml.js')
var outFile = './Output/result.codeml'
base.call_(process.argv[2], outFile, process.argv, codeml.run_)Basic usage: node app.js input.cnt [all parameters set by cnt file]
node app.js test.cnt ProtTest3
Run ProtTest3 program
var base = require('../../../Wrapper_Core/base-wrap')
var prottest = require('./prottest')
base.call_(process.argv[2], process.argv[3], process.argv, prottest.run_)Basic usage: node app.js path-to-jar input-file [insert any flags (from flags below)]
node app.js /path-to-jar/prottest-3.4.2.jar alignment -all-matrices -all-distributions -o example.txt| FLAG | DETAILS |
| --------------------- |:-------------------------------------------------:|
| -i | alignment_filename |
| -t | tree_filename (optional) |
| -o | output_filename (optional) |
| -[matrix] | Include matrix (Amino-acid) |
| -I | models with a proportion of invariable sites |
| -G | rate variation among sites and categories |
| -IG | models with both +I and +G |
| -all-distributions | rate variation among sites, categories and both |
| -ncat | number of categories |
| -F | models with empirical frequency estimation |
| -AIC | Akaike Information Criterion |
| -BIC | Bayesian Information Criterion |
| -AICC | Corrected Akaike Information Criterion |
| -DT | Decision Theory Criterion |
| -all | 7-framework comparison table |
| -S | Optimization strategy mode: [default: 0] |
| -s | Tree search operation for ML search |
| -t1 | Display best-model's newick tree |
| -t2 | Display best-model's ASCII tree |
| -tc | Display consensus tree with specified threshold |
| -threads | Number of threads requested to compute |
| -verbose | Verbose mode [default: false] |
Note: must have Java Runtime environment and ProtTest3 jar
jModelTest2
Run jModelTest2 program
var base = require('../../../Wrapper_Core/base-wrap')
var jmodeltest2 = require('./jmodeltest2')
base.call_(process.argv[2], process.argv[3], process.argv, jmodeltest2.run_)Basic usage: node app.js path-to-jar input-file -o output-file [insert any flags (from flags below)]
node app.js /path-to-jar/jModelTest.jar aP6.fas -o Output/Results.txt -f -i -g 4 -s 11 -AIC -a| FLAG | DETAILS |
| ------------------|:----------------------------------------------------------------------:|
| -a | Estimate model-averaged phylogeny for each active criterion |
| -t | Base tree for likelihood calculations (e.g., -t BIONJ) |
| -o | outputFile |
| -i | Include models with a proportion invariable sites |
| -machinesfile | Gets the processors per host from a machines file |
| -g | numberOfRateCategories |
| -getPhylip | Converts the input file into phylip format and exits |
| -G | threshold |
| -h | confidenceInterval |
| -AIC | Akaike Information Criterion |
| -BIC | Bayesian Information Criterion |
| -AICc | Corrected Akaike Information Criterion |
| -hLRT | Perform hierarchical likelihood ratio tests |
| -DT | Calculate the decision theory criterion |
| -f | Include models with unequals base frecuencies |
| -H | Information criterion for clustering search |
| -n | logSuffix |
| -O | Sets the hypothesis order for the hLRTs |
| -p | Calculate the parameter importances |
| -tr | Number of threads requested to compute |
| -v | Do model averaging and parameter importances |
| -s | Sets the number of substitution schemes |
| -u | treefile |
| -uLNL | Calculate delta AIC,AICc,BIC against unconstrained likelihood|
| -w | Prints out the PAUP block |
| -z | Strict consensus type for model-averaged phylogeny |
Note: must have Java Runtime environment and jModelTest2 jar
Pipes
Commands can be chained in series to pipe data between applications:
var shell = require('./phylo-node_pipes')Pipes dir contains the module for piping as well as example files. To execute example:
node pipe_example.jspipe_example.js pipes the output from an NCBI fetch API call into the alignment software MUSCLE and aligns the DNA using default settings
Note: must have MUSCLE in $PATH for pipe example
Testing
phylo-node was successfully tested on:
- [x] Microsoft Windows 7 Enterprise ver.6.1
- [x] MacOSX El Capitan ver.10.11.5
- [x] Linux Ubuntu 64-bit ver.14.04 LTS
To perform tests:
npm testTo ensure all developmental dependencies are installed:
npm install --devNote: if you get a permission error when runnning tests you may have to chmod mocha
chmod 0777 mochaDocumentation
Skinner M.E., Uzilov A.V., Stein L.D., Mungall C.J., Holmes I.H. (2009). JBrowse: a next-generation genome browser. Genome Research, 19(9):1630-1638
Langmead B, Salzberg S. (2012). Fast gapped-read alignment with Bowtie 2. Nature Methods, 4;9(4):357-9
Bolger, A. M., Lohse, M., & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics, btu170
Guindon S., Dufayard J.F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology, 59(3):307-21
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M and Rozen SG. (2012). Primer3 - new capabilities and interfaces. Nucleic Acids Res. 40(15):e115
Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5):1792-1797
Edgar, R.C. (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics, (5)113
Sievers F, Wilm A, Dineen DG, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology 7:539
Lassmann T, Sonnhammer EL. (2005). Kalign--an accurate and fast multiple sequence alignment algorithm. BMC Bioinformatics. 12;6:298
Suyama M, Torrents D, Bork P (2006). PAL2NAL: robust conversion of protein sequence alignment into the corresponding codon alignments. Nucleic Acids Res. 34:W609-W612
Massingham T, Goldman N (2005) Detecting amino acid sites under positive selection and purifying selection. Genetics 169: 1853-1762
Yang, Z (2007) PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586-91.
Yang, Z (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 13(5):555-6
Darriba D, Taboada GL, Doallo R, Posada D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics, 27:1164-1165
Darriba D, Taboada GL, Doallo R, Posada D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9(8), 772
Contributing
All contributions are welcome.
Support
If you have any problem or suggestion please open an issue here.
License
The MIT License
Copyright (c) 2016, dohalloran
Permission is hereby granted, free of charge, to any person obtaining a copy of this software and associated documentation files (the "Software"), to deal in the Software without restriction, including without limitation the rights to use, copy, modify, merge, publish, distribute, sublicense, and/or sell copies of the Software, and to permit persons to whom the Software is furnished to do so, subject to the following conditions:
The above copyright notice and this permission notice shall be included in all copies or substantial portions of the Software.
THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY, FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM, OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE SOFTWARE.